CASOS CLÍNICOS
Aniridia familiar por deleción reguladora de PAX6: desafíos diagnósticos y relevancia del mosaicismo materno
Mayra S. Arbeloa, María G. Obregona, María C. Mansillab, Jean M. Rozetc, Lucas Fares-Taiec, Julie Plaisanciéd,Victoria Huckstadta
a Servicio de Genética, Hospital de Pediatría J. P. Garrahan, Buenos Aires, Argentina.
b Servicio de Oftalmología, Hospital de Pediatría J. P. Garrahan, Buenos Aires, Argentina.
c Laboratorio de Genética en Oftalmología (LGO), INSERM UMR 1163 e Instituto Imagine de Enfermedades Genéticas y Universidad Descartes, 75015 París, Francia.
d Genetista del Centro de Referencia de Enfermedades Raras en Genética Oftalmológica, Toulouse, Francia.
Recibido: 11 de agosto de 2025.
Aprobado: 7 de noviembre de 2025.
Autor corresponsal
Dra. Mayra S. Arbelo
Servicio de Genética
Hospital de Pediatría J. P. Garrahan
Combate de los Pozos 1881
(C1245) Ciudad de Buenos Aires.
Argentina
+54 11 4122-6000
arbelomayra.s@gmail.com
Oftalmol Clin Exp (ISSNe 1851-2658)
2025; 18(4): e542-e547.
DOI: https://doi.org/10.70313/2718.7446.v18.n4.469
Resumen
Objetivo: La aniridia es un trastorno panocular poco frecuente que puede heredarse de forma autosómica dominante y en el 90% de los casos puede ser causada por variantes de nucleótido único en el gen PAX6, deleciones que afectan al gen completo o a la región regulatoria 11p13, o por compromiso de genes contiguos. Cerca del 30% de las variantes es de novo y la tasa de mosaicismo es cercana al 17,5%. Nuestro objetivo es reportar y caracterizar un caso familiar de aniridia asociada a una microdeleción en la región reguladora de PAX6, evaluando su correlación genotipo-fenotipo y la contribución del mosaicismo materno al espectro clínico.
Caso clínico: Presentamos una paciente de 12 años con diagnóstico posnatal de aniridia bilateral con padres referidos sanos. Se realizó un panel de genes asociado a patología ocular sin variantes relevantes y un a-CGH (180k) que reportó una deleción de 657 kb en 11p13 que comprende 4 genes OMIM vinculados con la región regulatoria de PAX6. El estudio de a-CGH en su madre reportó la misma microdeleción en mosaico (20%), detectándose posteriormente aniridia parcial y catarata nuclear en ojo izquierdo.
Conclusión: Describimos un caso familiar de aniridia con herencia autosómica dominante causada por una deleción en la región reguladora del gen PAX6, con menor expresión en su madre por la presencia del mosaico. Debido a la heterogeneidad clínica, genética y la expresión variable involucradas, destacamos la importancia de estudiar a los pacientes combinando diferentes técnicas moleculares para un adecuado diagnóstico y asesoramiento genético.
Palabras clave: aniridia, array CGH, mosaicismo, PAX6.
Familial aniridia due to a regulatory PAX6 deletion: diagnostic challenges and the relevance of maternal mosaicism
Abstract
Objective: Aniridia is a rare panocular disorder. It is inherited in an autosomal dominant manner and in 90% of cases is caused by variants in PAX6 which can be single nucleotide variants, deletions affecting the entire gene, or the regulatory region 11p13, or comprise contiguous gene syndromes. Up to 30% percent of variants are de novo, and the rate of mosaicism could reach 17.5%. Our objective was to report and characterize a familial case of aniridia associated with a microdeletion in the regulatory region of PAX6, assessing its genotype-phenotype correlation and the contribution of maternal mosaicism to the clinical spectrum.
Case report: We present a 12-year-old patient with postnatal diagnosis of bilateral aniridia. A gene panel for ocular pathology that did not detect any variants relevant and an a-CGH (180k), which reported a 657 kb deletion in 11p13 comprising 4 OMIM genes linked to the regulatory region of PAX6. A-CGH study in the mother reported the same microdeletion in mosaic (20%), with nuclear cataract in the left eye and partial aniridia.
Conclusions: We describe a case of familial aniridia with autosomal dominant inheritance, caused by deletion of the PAX6 gene regulatory region, with the mother exhibiting milder expression due to the deletion being a mosaic. Because of the clinical and genetic heterogeneity, and variable expression involved, we emphasize the importance of molecular analysis of patients combining different laboratory tools for adequate diagnosis and genetic counseling.
Keywords: aniridia, array CGH, mosaicism, PAX6.
Aniridia familiar devido à deleção regulatória de PAX6: desafios diagnósticos e relevância do mosaicismo materno
Resumo
Objetivo: A aniridia é uma doença panocular rara que pode ser herdada de forma autossômica dominante e, em 90% dos casos, pode ser causada por variantes de nucleotídeo único no gene PAX6, deleções que afetam todo o gene ou a região regulatória 11p13, ou pelo envolvimento de genes contíguos. Aproximadamente 30% das variantes são de novo, e a taxa de mosaicismo é próxima de 17,5%. Nosso objetivo é relatar e caracterizar um caso familiar de aniridia associado a uma microdeleção na região regulatória do PAX6, avaliando sua correlação genótipo-fenótipo e a contribuição do mosaicismo materno para o espectro clínico.
Caso clínico: Apresentamos o caso de uma paciente de 12 anos com diagnóstico pós-natal de aniridia bilateral, cujos pais foram relatados como saudáveis.
Foi realizado um painel de genes associados à patologia ocular, sem que variantes relevantes fossem encontradas, e uma análise a-CGH (180k) revelou uma deleção de 657 kb em 11p13, compreendendo quatro genes OMIM ligados à região regulatória do PAX6. A análise de a-CGH da mãe revelou a mesma microdeleção em mosaico (20%), e ela foi posteriormente diagnosticada com aniridia parcial e catarata nuclear no olho esquerdo.
Conclusão: Descrevemos um caso familiar de aniridia com herança autossômica dominante causada por uma deleção na região regulatória do gene PAX6, com menor expressão na mãe devido à presença de mosaicismo. Dada a heterogeneidade clínica e genética e a expressão gênica variável envolvida, enfatizamos a importância do estudo dos pacientes utilizando uma combinação de diferentes técnicas moleculares para um diagnóstico preciso e aconselhamento genético adequado.
Palavras-chave: aniridia, array CGH, mosaicismo, PAX6.
Introducción
La aniridia es un trastorno panocular poco frecuente, caracterizado por un grado variable de hipoplasia o ausencia del iris, aumento progresivo de la presión intraocular que provoca glaucoma, afección del cristalino (catarata y subluxación del cristalino), la fóvea y el nervio óptico generando hipoplasia y coloboma. Presenta además otras anomalías oculares como estrabismo, microftalmía, nistagmus con deterioro progresivo de la agudeza visual. Puede ocurrir en forma aislada y heredarse de forma autosómica dominante1, con alta penetrancia y expresividad variable o como parte del síndrome de WAGR (tumor de Wilms, aniridia, anomalías genitales y retraso del neurodesarrollo)2.
Es decir, podemos encontrarnos con formas leves de aniridia con cambios sutiles en la arquitectura del iris, buena visión y estructura foveal normal hasta casos más graves con pérdida de la agudeza visual y microftalmía severa2.
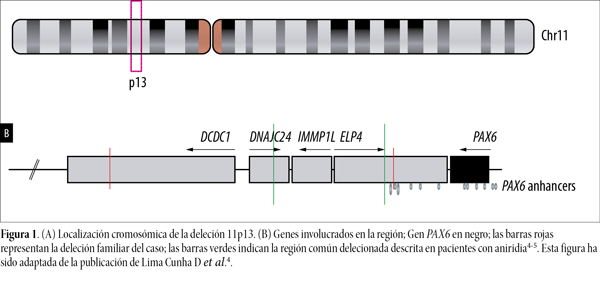
Con el avance de las técnicas de diagnóstico molecular se logra el diagnóstico genético en aproximadamente el 90% de los pacientes. Dos tercios de los casos son causados por variantes puntuales en el gen PAX6 o deleciones que afectan al gen completo. En el tercio restante son ocasionados por afectación de la región reguladora 11p13 que controla su expresión (con los genes ELP4, DPH4, DCDC1, DCDC5, MPPED2 e IMMP1L) o deleciones de genes contiguos a PAX6, especialmente WT1 (fig. 1)3.
El 70% de los pacientes posee un progenitor afectado y el 30% son casos de novo. Se describe que la tasa de mosaicismo en los padres afectados podría llegar al 17,5%, pudiendo causar fenotipos oculares más leves en comparación con su descendencia, pasando inadvertido a lo largo de la vida, dificultando su diagnóstico, el que se suele realizar en la adultez cuando aparece un descendiente afectado con mayor severidad4. En este contexto, nuestro propósito ha sido informar y caracterizar un caso familiar de aniridia asociada a una microdeleción en la región reguladora de PAX6, evaluando su correlación genotipo-fenotipo y la contribución del mosaicismo materno al espectro clínico.
Caso clínico
Paciente evaluada por primera vez en nuestro hospital (Hospital de Pediatría J. P. Garrahan, ciudad de Buenos Aires, Argentina) a los 9 años por presentar anomalía ocular. Es la única hija de una pareja referida sana, no consanguínea, sin antecedentes familiares de relevancia. Se había realizado diagnóstico de glaucoma congénito al nacimiento y aniridia bilateral tras realizar estudio de fondo de ojos en un control de rutina; requirió cirugía de glaucoma a los 11 días de vida en otra institución.
Al examen oftalmológico se observó: niña sin nistagmus. La agudeza visual mejor corregida fue de 2 décimas y 7 décimas, la presión intraocular de 19 mmHg en ambos ojos. La refracción bajo cicloplejía fue en ojo derecho de esférico +2,5 combinado con cilindro +2,5 a 115° y ojo izquierdo esférico +2 combinado con cilindro +1,5 a 123°.
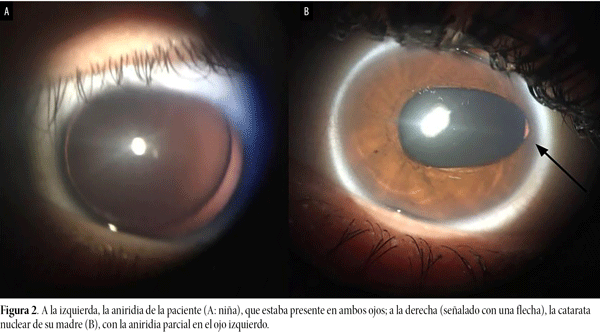
En el fondo de ojo del ojo derecho se observó mácula y papila dentro de parámetros normales con árbol vascular normal e hipertrofia del epitelio pigmentario de retina; mientras que la del ojo izquierdo mostró mácula y papila dentro de parámetros normales con árbol vascular normal sin otras lesiones (fig. 2A).
En la primera consulta se realizó cariotipo con bandeo G y luego panel para genes asociados a patología ocular que fueron normales. Se completó el estudio con a-CGH (180k), que informó: arr[GRCh37]11p13(31024178_31682022)x1 deleción de 657 kb que comprende 4 genes OMIM: DCDC1, DNAJC24, IMMP1L y ELP4 vinculados con la región reguladora de PAX6 (que incluye elementos reguladores en intrones del gen y en regiones intergénicas ubicadas antes y después de él. Sus elementos son cruciales para la expresión de PAX6 durante el desarrollo de diversas estructuras oculares). El estudio de a-CGH en la madre informó la misma deleción en mosaico (25-25%) (arr[GRCh37]11p13(31024178_31682022)x1~2) (fig. 1B). El padre no estaba disponible para el estudio. Debido a los resultados obtenidos en el estudio molecular, se realizó examen ocular a su madre y se identificó en el ojo izquierdo aniridia parcial con catarata nuclear (fig. 2B).
Discusión
El caso presentado ilustra de manera clara la complejidad genética y fenotípica asociada a la aniridia, un trastorno panocular cuya etiología está estrechamente vinculada con alteraciones del gen PAX6 y de sus regiones reguladoras. En nuestro caso, la identificación de una microdeleción en 11p13 permitió establecer con precisión el mecanismo patogénico responsable, evidenciando un patrón de herencia autosómica dominante con expresividad variable. Esta variabilidad se hizo particularmente evidente en la madre —portadora en mosaico de la misma deleción— en quien la presentación clínica fue más leve y parcialmente inadvertida hasta la realización del estudio molecular. Este hallazgo coincide con lo reportado en la literatura, donde el mosaicismo somático o germinal se reconoce cada vez más como una causa relevante de fenotipos atenuados en enfermedades monogénicas.
La reevaluación diagnóstica derivada del estudio molecular tuvo un impacto directo en el asesoramiento genético. Mientras que inicialmente la paciente había sido interpretada como un caso de aniridia de novo, el hallazgo de la variante familiar obliga a reclasificarla como una forma hereditaria con un riesgo de recurrencia de hasta el 50%. Esta diferencia no es trivial: redefine la información transmitida a la familia, modifica la estimación del riesgo reproductivo y permite anticipar la posible presencia de manifestaciones oculares en otros miembros, incluso en aquellos con fenotipos sutiles. En este sentido, el reconocimiento del mosaicismo materno es clave, ya que los mosaicismos de baja proporción pueden pasar inadvertidos en la evaluación clínica pero mantienen capacidad de transmisión germinal plena.
El caso también subraya la importancia de adoptar un abordaje diagnóstico integral mediante el uso complementario de distintas herramientas moleculares. La combinación de paneles dirigidos de genes, técnicas para detección de CNVs (como a-CGH o MLPA) y metodologías de NGS (paneles ampliados o exoma) permite alcanzar una mayor sensibilidad diagnóstica6. En particular, las deleciones en regiones reguladoras de PAX6 pueden no detectarse mediante secuenciación estándar, por lo que el empleo de técnicas específicas para identificar variaciones estructurales resulta imprescindible. La literatura actual subraya que entre un 10% y un 15% de los casos clínicamente compatibles con aniridia presentan alteraciones en regiones no codificantes, lo que refuerza la necesidad de incluir sistemáticamente estas metodologías en la evaluación6.
Otro aspecto relevante es la correlación genotipo-fenotipo. Las microdeleciones que afectan regiones regulatorias de PAX6 suelen asociarse con un espectro clínico más amplio, que puede incluir aniridia completa, parcial o manifestaciones asociadas como catarata, opacidades corneales o alteraciones foveales. La presentación más leve observada en la madre es consistente con esta variación, exacerbada en este caso por la condición de mosaicismo. Estos hallazgos nos recuerdan que la fenotipificación detallada de los familiares es esencial y que manifestaciones sutiles como hipoplasia foveal leve o cataratas nucleares incipientes pueden constituir pistas diagnósticas de un trastorno genético subyacente.
Finalmente, este caso refuerza la recomendación de realizar estudios clínicos y moleculares completos a ambos padres en todo paciente con diagnóstico de aniridia, independientemente de la apariencia fenotípica inicial. La identificación de mosaicismos o variantes heredadas modifica sustancialmente el asesoramiento genético y optimiza la planificación reproductiva. En un contexto más amplio, nuestro reporte contribuye a visibilizar la importancia del análisis molecular integral y la evaluación familiar ampliada en enfermedades oculares hereditarias, especialmente aquellas asociadas a reguladores clave del desarrollo ocular como PAX6.
Conclusión
Este caso familiar de aniridia demuestra cómo las microdeleciones en la región reguladora de PAX6 pueden generar un espectro clínico variable, especialmente en presencia de mosaicismo, que atenúa la expresión fenotípica y dificulta el reconocimiento clínico en los progenitores. La identificación de la deleción en ambos miembros y la detección del mosaicismo materno permitieron reclasificar el caso como una aniridia autosómica dominante, modificando de manera sustancial el asesoramiento genético y el cálculo del riesgo de recurrencia. Estos hallazgos refuerzan la necesidad de emplear estrategias moleculares complementarias —incluyendo secuenciación, análisis de CNVs y herramientas basadas en NGS— así como de evaluar exhaustivamente a ambos padres en todos los casos de aniridia a fin de asegurar un diagnóstico preciso y una adecuada orientación clínica y reproductiva.
Referencias
-
Cheng F, Song W, Kang Y, Yu S, Yuan H.
A 556 kb deletion in the downstream region of the PAX6 gene causes familial aniridia and other eye anomalies in a Chinese family.
Mol Vis. 2011;17:448–455.
http://www.molvis.org/molvis/v17/a50/
-
Moosajee M, Hingorani M, Moore AT.
PAX6-related aniridia.
2003 May 20 [updated 2018 Oct 18].
In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors.
GeneReviews® [Internet].
Seattle (WA): University of Washington; 1993–2025.
https://www.ncbi.nlm.nih.gov/books/NBK1360/
-
Li Y, Chen J, Zheng Y, Chen Z, Wang T, Sun Q, Wan X, Liu H, Sun X.
A novel microdeletion of 517 kb downstream of the PAX6 gene in a Chinese family with congenital aniridia.
BMC Ophthalmol. 2023;23(1):393.
https://doi.org/10.1186/s12886-023-03147-1
-
Lima Cunha D, Arno G, Corton M, Moosajee M.
The spectrum of PAX6 mutations and genotype–phenotype correlations in the eye.
Genes (Basel). 2019;10(12):1050.
https://doi.org/10.3390/genes10121050
-
Wawrocka A, Krawczynski MR.
The genetics of aniridia: simple things become complicated.
J Appl Genet. 2018;59(2):151–159.
https://doi.org/10.1007/s13353-017-0426-1
-
Vasilyeva TA, Voskresenskaya AA, Käsmann-Kellner B, Khlebnikova OV, Pozdeyeva NA, Bayazutdinova GM, Kutsev SI, Ginter EK, Semina EV, Marakhonov AV, Zinchenko RA.
Molecular analysis of patients with aniridia in the Russian Federation broadens the spectrum of PAX6 mutations.
Clin Genet. 2017;92(6):639–644.
https://doi.org/10.1111/cge.13019
LEYENDAS
Figura 1. (A) Localización cromosómica de la deleción 11p13. (B) Genes involucrados en la región; Gen PAX6 en negro; las barras rojas representan la deleción familiar del caso; las barras verdes indican la región común delecionada descrita en pacientes con aniridia4-5. Esta figura ha sido adaptada de la publicación de Lima Cunha D et al.4.
Figura 2. A la izquierda, la aniridia de la paciente (A: niña), que estaba presente en ambos ojos; a la derecha (señalado con una flecha), la catarata nuclear de su madre (B), con la aniridia parcial en el ojo izquierdo.