CASOS CLÍNICOS
Sophia Arias, Estefanía Lis Luque, Sofia Inés Aliaga, Carolina Fernanda Pastorello, Pablo Francisco Colom, Emiliano Facundo Ross
Recibido: 6 de junio de 2025.
Aprobado: 2 de octubre de 2025.
Autor corresponsal
Dra. Sophia Arias
Hospital Nacional Prof. Alejandro Posadas
(1684) El Palomar, Provincia de Buenos Aires
Argentina.
+54 11 4469-9300
sophiaaarias@gmail.com
Oftalmol Clin Exp (ISSNe 1851-2658)
2025; 18(4): e487-e493.
DOI: https://doi.org/10.70313/2718.7446.v18.n4.456
Retinitis pigmentosa in patients with mucopolysaccharidosis II: report of two cases
Abstract
Purpose: To highlight the importance of regular ophthalmologic follow-up in patients diagnosed with mucopolysaccharidosis type II (MPS II) for the early detection of associated ocular conditions, particularly retinitis pigmentosa (RP), through the description of two clinical cases.
Case report: MPS II is a lysosomal storage disorder caused by mutations in the IDS gene (Xq28), inherited in an X-linked manner, leading to iduronate-2-sulfatase deficiency and accumulation of glycosaminoglycans in multiple organs. Ocular manifestations may include corneal opacities, macular dysfunction, optic neuropathy, and retinal pigmentary changes. RP represents a heterogeneous group of inherited dystrophies characterized by progressive photoreceptor degeneration, resulting in visual field constriction and macular involvement at advanced stages.
Our two MPS II patients presented with decreased visual acuity. Fundus examination and electrophysiological studies revealed findings consistent with RP, confirming the coexistence of both conditions.
Conclusion: Regular ophthalmologic evaluation in patients with MPS II is essential for the early detection of retinal complications such as RP, allowing an interdisciplinary approach and proper follow-up to help preserve visual function and improve quality of life.
Keywords: mucopolysaccharidosis II, retinitis pigmentosa, retinal dystrophies, lysosomal storage diseases.
Retinite pigmentosa em pacientes com mucopolissacaridose tipo II: relato de dois casos
Resumo
Objetivo: Destacar a importância do acompanhamento oftalmológico periódico em pacientes diagnosticados com mucopolissacaridose tipo II (MPS II) para a detecção precoce de doenças oculares associadas —em particular, retinose pigmentar— por meio da descrição de dois casos clínicos.
Caso clínico: A mucopolissacaridose tipo II é uma doença de armazenamento lisossômico causada por mutações no gene IDS (Xq28), herdadas no cromossomo X, que levam à deficiência da enzima iduronato-2-sulfatase e ao acúmulo de glicosaminoglicanos em diversos órgãos. Suas manifestações oculares incluem opacidades da córnea, disfunção macular, neuropatia óptica e alterações pigmentares da retina. A retinose pigmentar, por outro lado, compreende um grupo heterogêneo de distrofias hereditárias caracterizadas pela degeneração progressiva dos fotorreceptores com redução do campo visual e envolvimento macular em estágios avançados.
Dois pacientes com mucopolissacaridose tipo II apresentaram diminuição da acuidade visual. O exame de fundo de olho e os estudos eletrofisiológicos foram compatíveis com retinose pigmentar, confirmando a coexistência de ambas as condições.
Conclusão: O exame oftalmológico regular em pacientes com mucopolissacaridose tipo II é essencial para a detecção precoce de complicações retinianas, como a retinose pigmentar, permitindo uma abordagem interdisciplinar e um acompanhamento adequado que contribui para a preservação da função visual e a melhoria da qualidade de vida.
Palavras-chave: mucopolissacaridose II, retinose pigmentar, distrofias da retina, doenças de armazenamento lisossômico.
Introducción
Las mucopolisacaridosis (MPS) son un grupo heterogéneo de trastornos caracterizados por la acumulación generalizada de glucosaminoglucanos (GAG) tanto a nivel sistémico como en los tejidos oculares1.
La mucopolisacaridosis tipo II (MPS II) o síndrome de Hunter es una enfermedad genética rara, con una incidencia que oscila entre 0,38 y 1,09 por 100.000 varones nacidos vivos y es la única mucopolisacaridosis hereditaria ligada al cromosoma X. La causa una deficiencia en la enzima iduronato-2-sulfatasa (IDS)2. Entre las manifestaciones oftalmológicas se han descrito opacidades corneales, anomalías retinales, maculares y afectación del nervio óptico3-6.
En estudios previos se ha señalado que en fenotipos leves —con ausencia de alteraciones cognitivas— se han observado hallazgos similares a los reportados en este trabajo, los que son compatibles con la presencia de retinosis pigmentaria7-8.
La retinosis pigmentaria (RP) es una distrofia hereditaria caracterizada por degeneración progresiva de los fotorreceptores y disfunción del epitelio pigmentario de la retina. Aunque la afectación ocular es común en la MPS II, su asociación con RP no ha sido ampliamente estudiada6-9.
Autores como Kowalsky y colaboradores reportaron la presencia de grandes cantidades de heparán sulfato en el epitelio pigmentario de la retina, en la capa de células ganglionares y en la capa de fibras nerviosas10. Además, se ha descrito la aglomeración bilateral y simétrica en la cabeza del nervio óptico en pacientes con MPS II, probablemente debido a la deposición de GAG en el espacio subaracnoideo, lo que lleva a la compresión del nervio óptico10-13.
Por lo tanto, el objetivo es destacar la importancia del seguimiento oftalmológico en esta población. Esto nos permitirá la detección oportuna de enfermedades asociadas, como la RP mediante un examen clínico ocular y el uso criterioso de estudios complementarios.
Casos clínicos
A continuación se presentan dos casos de pacientes con MPS II que presentan hallazgos compatibles con RP.
Caso 1
El primer caso es sobre un paciente masculino de 27 años con diagnóstico de MPS II a los 7 años sin alteraciones en su desarrollo intelectual. Al examen oftalmológico presentó: agudeza visual sin corrección (AVSC) ojo derecho (OD) 3/10, ojo izquierdo (OI) 7/10. Su agudeza visual mejor corregida (AVMC) (OD +1,50 +1,50 x 150° / OI +2,00 +1,50 x 50°) fue 8/10 ambos ojos (AO). En la biomicroscopía (BMC) se encontró la córnea transparente en AO, resto sin particularidades. Al fondo de ojos: medios transparentes AO, papila rosada de bordes netos y definidos, alteración del brillo macular bilateral, afinamiento vascular, presencia de aglomerados de pigmento con apariencia de “espículas óseas” en retina periférica 360 grados, retina aplicada en AO. Hallazgos sugerentes de retinosis pigmentaria en AO.
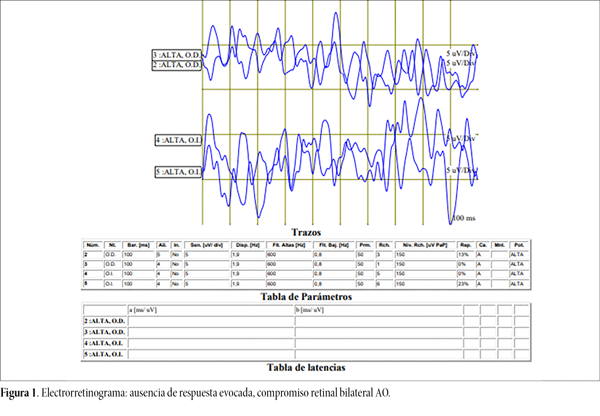
Ante la sospecha de RP se solicitó: evaluación genética para confirmar el diagnóstico, que al día de la fecha se encuentra pendiente. Electrorretinograma que informa: ausencia de respuesta evocada AO, concluyendo en compromiso retinal bilateral (fig. 1).
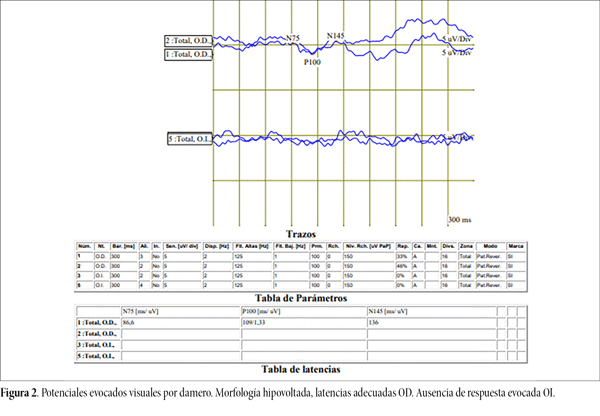
Potenciales evocados visuales (PEV) por damero que informa que OD presenta respuesta de morfología hipovoltada y latencias adecuadas (fig. 2). Buen replicado OI ausencia de respuesta evocada. Concluyendo en compromiso de la vía visual derecha. Y PEV por flash que informa respuesta evocada de morfología hipovoltada y latencias adecuadas. Buen replicado en AO, concluyendo permeable a la luz.
Caso 2
El segundo paciente es un masculino de 29 años con diagnóstico de MPS II en la infancia sin alteraciones en su desarrollo intelectual. En 2018 se realizó determinación del gen de IDS por método molecular. Al momento de la consulta se encontraba en tratamiento con terapia de reemplazo enzimático que había comenzado en el año 2015.
Al examen oftalmológico presentó una AVSC 4/10 AO. Mientras que la AVMC fue de (OD +3,25 -1,00 x 60 / OI +3,25 -1,50 x 125) 8/10 AO. En la BMC sin particularidades en AO.
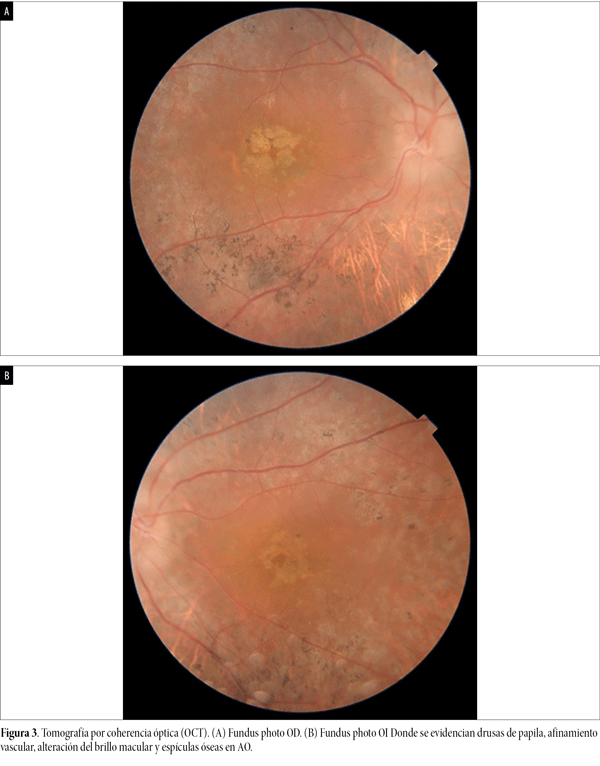
En el fondo de ojos se constataron medios transparentes AO (fig. 3). Papila de bordes poco definidos compatible con drusas de papila confirmada por ecografía ocular AO. Alteración del brillo macular bilateral, vasos de calibre conservado, presencia de aglomerados de pigmento con apariencia de “espículas óseas” en retina periférica 360 grados con retina aplicada en AO.
Al momento de la consulta acudió con resultado de PEV solicitado previamente, donde se informaba que no se había obtenido oscilaciones mensurables postestimulación bilateral, por lo que se concluye ERG abolido bilateral. Compatible con hallazgos sugerentes de retinosis pigmentaria en AO.
Discusión
Los pacientes con MPS II pueden desarrollar depósitos de GAG en la retina, lo que provoca alteraciones en el epitelio pigmentario de la retina y disfunción en los fotorreceptores, factores que podrían contribuir al desarrollo de RP. Las anormalidades retinales parecen ser una característica común en los pacientes con MPS II. Este proceso a menudo se manifiesta tempranamente, incluso antes del diagnóstico de MPS II, lo que sugiere que la retinopatía podría ser una manifestación primaria de la enfermedad2-7, 12-13.
En nuestros pacientes, los signos sugestivos de RP son la presencia de espículas óseas en 360° en la retina periférica junto con alteraciones en el brillo macular. Asimismo, este patrón de signos se ha identificado con frecuencia en estudios de pacientes con MPS II11-13.
La evaluación genética podría ser una herramienta útil para determinar si la retinosis pigmentaria es una manifestación secundaria de la MPS II o si, por el contrario, existen mutaciones en genes que afectan ambos trastornos de manera independiente. Profundizar en esta relación podría tener implicancia en el diagnóstico y tratamiento de estas patologías
Conclusión
Se subraya la importancia del examen oftalmológico protocolizado en estos pacientes. En ambos casos, los hallazgos clínicos y electrofisiológicos fueron consistentes con un diagnóstico de RP. Este seguimiento permite la detección oportuna de complicaciones visuales severas y resalta la necesidad de considerar la RP como una manifestación asociada a esta enfermedad. La evaluación genética podría ser útil para diferenciar si la RP es una manifestación secundaria de la MPS II o si mutaciones genéticas independientes lo que podría abrir nuevas oportunidades para la investigación.
Referencias
LEYENDAS
Figura 1. Electrorretinograma: ausencia de respuesta evocada, compromiso retinal bilateral AO.
Figura 2. Potenciales evocados visuales por damero. Morfología hipovoltada, latencias adecuadas OD. Ausencia de respuesta evocada OI.
SEBAS EN LA SIGUIENTE FIGURA PONÉ A y B:
Figura 3. Tomografía por coherencia óptica (OCT). (A) Fundus photo OD. (B) Fundus photo OI Donde se evidencian drusas de papila, afinamiento vascular, alteración del brillo macular y espículas óseas en AO.