
Figura 1. Segmento anterior del OI con córnea transparente, imagen de cobre martillado en el endotelio, pupila corectópica, atrofia del iris difusa y ectropión uveal.
REPORTE DE CASO
Síndrome endotelial iridocorneal: reporte de un caso clínico
Guillermo Raúl Vera-Duarte, Martín Fernando Arrúa, Luis B. González-Sanabria, Diógenes O. Cibils-Farres
Servicio de Oftalmología, Hospital de Clínicas, Universidad Nacional de Asunción, Paraguay.
Recibido: 19 de julio de 2021.
Aprobado: 20 de noviembre de 2021.
Autor corresponsal
Dr. Guillermo R. Vera-Duarte
Coronel Cazal, San Lorenzo
Paraguay
595 (21) 585 730
guillermoveraduarte@gmail.com
Oftalmol Clin Exp (ISSN 2718-7446)
2022; 15(1): e79-e84.
Resumen
Objetivo: Presentar el caso clínico de una paciente con síndrome del endotelio irido-corneal (ICE).
Caso clínico: Paciente de sexo femenino de 42 años con antecedente de disminución de la agudeza visual (AV) en ojo izquierdo (OI). Tenía AV mejor corregida en ojo derecho (OD) 10/10 y en OI, 8/10. Presión intraocular (PIO) en OD: 17 mmHg, OI 35 mmHg con terapia hipotensora máxima. En la gonioscopía se observó la presencia de sinequias anteriores periféricas altas y de base ancha insertadas a nivel de malla trabecular y línea de Schwalbe con cierre angular secundario. En tomografía de coherencia óptica se constató la pérdida de la capa de fibras nerviosas peripapilares en OI, sin alteraciones en OD. La campimetría computarizada mostró defectos arqueados en OI. En la microscopía especular se observó disminución en el recuento de células endoteliales con polimegatismo y pleomorfismo asociado, así como aumento del espesor corneal. Por lo anterior se estableció el diagnóstico de ICE y se realizó cirugía de implante valvular con dispositivo valvulado de Ahmed en OI. Al año, la PIO se mantiene en 12 mmHg con córnea trasparente.
Conclusión: El síndrome ICE presenta una proliferación endotelial corneal que migra hacia el ángulo iridocorneal y sobre el iris con diferentes grados de edema corneal, alteraciones iridianas y cierre angular progresivo con el consiguiente desarrollo de glaucoma. Ante la sospecha de un ICE es importante realizar un examen completo del segmento anterior buscando signos en el iris como en la córnea para realizar un correcto diagnóstico y su oportuno tratamiento.
Palabras clave: síndrome iridocórneo-endotelial, proliferación endotelial secundaria, glaucoma.
Iridocorneal endothelial syndrome: a clinical case report
Abstract
Objective: To present the case of a patient with iridocorneal endothelial (ICE) syndrome.
Clinical case: Forty-two-year-old female patient with a history of visual acuity (VA) loss in the left eye (LE) whose best spectacle-corrected VA was 10/10 in the right eye (RE) and 8/10 in the LE. Her intraocular pressure (IOP) was 17 mmHg in the RE and 35 mmHg in the LE with maximum IOP-lowering therapy. Gonioscopy revealed the presence of high, broad-based peripheral anterior synechiae inserted at the level of the trabecular meshwork and Schwalbe’s line, with secondary angle closure. Optical coherence tomography evidenced that there was peripapillary nerve fiber layer loss in the LE, though the RE had no alterations. The presence of arcuate visual field defects in the LE was found by computerized perimetry. With specular microscopy, a decrease in endothelial cell count, with associated polymegethism and pleomorphism, as well as increased corneal thickness, was observed. Diagnosis of ICE syndrome was made based on all these findings, and a surgical procedure was performed on the LE using the Ahmed valve implantation technique. One year after the procedure, the IOP remains in 12 mmHg with a transparent cornea.
Conclusion: In ICE syndrome there is corneal endothelial proliferation that migrates to the iridocorneal angle, and on the iris, with different levels of corneal edema, iris alterations and progressive angle closure, thus leading to the development of glaucoma. When ICE syndrome is suspected, it is important to examine the whole anterior segment thoroughly looking for any signs on the iris as well as on the cornea in order to make accurate diagnosis and to administer timely treatment.
Key words: iridocorneal endothelial syndrome, secondary endothelial proliferation, glaucoma.
Síndrome endotelial iridocorneana: um relato de caso clínico
Resumo
Objetivo: Apresentar o caso clínico de um paciente com síndrome endotelial iridocorneana (ICE).
Caso clínico: Uma paciente de 42 anos de idade com histórico de diminuição da acuidade visual (VA) no olho esquerdo (OE). Ela teve melhor correção de VA no olho direito (OD) 10/10 e no OE, 8/10. Pressão intra-ocular (PIO) em OD: 17 mmHg, OI 35 mmHg com terapia hipotensiva máxima. A gonioscopia mostrou a presença de sinéquias anteriores periféricas altas e amplas inseridas no nível da malha trabecular e da linha de Schwalbe com fechamento angular secundário. A tomografia de coerência óptica mostrou perda da camada de fibra nervosa peripapilar no OE, sem alterações no OD. A campimetria computadorizada mostrou defeitos arqueados no OE. A microscopia especular mostrou uma diminuição da contagem de células endoteliais com polimorfismo e pleomorfismo associado, bem como um aumento da espessura da córnea. Portanto, o diagnóstico da ICE foi estabelecido e a cirurgia de implantação de válvula foi realizada com um dispositivo valvular Ahmed no OI. Em um ano, a PIO permaneceu em 12 mmHg com uma córnea transparente.
Conclusão: A síndrome ICE apresenta uma proliferação endotelial corneana que migra para o ângulo iridocorneano e sobre a íris com diferentes graus de edema corneano, alterações da íris e fechamento angular progressivo com o conseqüente desenvolvimento de glaucoma. Se houver suspeita do ICE, é importante realizar um exame completo do segmento anterior procurando sinais na íris e na córnea, a fim de fazer um diagnóstico correto e um tratamento oportuno.
Palavras-chave: síndrome endotelial iridocorneana, ICE, proliferação endotelial secundária, glaucoma.
INTORUCCIÓN
El síndrome del endotelio iridocorneal (ICE, según sus siglas en inglés) constituye un grupo de afecciones oculares que tienen en común la proliferación anormal de células endoteliales corneales que migran hacia el ángulo iridocorneal y sobre el iris1. Se considera una enfermedad no hereditaria, frecuentemente unilateral y progresiva1-3. El síndrome de ICE se asocia con frecuencia a glaucoma, ya sea por obstrucción del ángulo de las células en proliferación o por contracción de su membrana basal sobre el iris, que tira del iris periféricamente y tienden a formar sinequias anteriores periféricas (PAS). La proliferación de estas células también predispone al edema y descompensación corneal2, 4.
El ICE se clasifica en el síndrome del nevo del iris (Cogan-Reese), el síndrome de Chandler (CS) y la atrofia esencial progresiva del iris (API)1-3; la evidencia patológica y clínica sugiere que todas estas variedades representan una continua manifestación clínica debido al proceso patológico que implica la proliferación anormal de las células endoteliales corneales4.
Este síndrome es poco frecuente, sin embargo es importante su conocimiento debido a que se encuentra relacionado con glaucoma de difícil manejo debido principalmente a las sinequias anteriores periféricas y/o por el recubrimiento de la malla trabecular de la membrana celular, característico de esta afección3. Presenta predilección por el sexo femenino y no existe una asociación clara con alguna enfermedad sistémica1-3.
La clínica frecuente de estos pacientes suele ser la de disminución de la agudeza visual (AV) y puede ir acompañada de dolor2. La disminución de la AV empeora por las mañanas debido a la menor oxigenación corneal durante el cierre palpebral nocturno que va mejorando durante el transcurso del día2. Un diagnóstico temprano es útil para su mejor manejo porque su progresión constituye un desafío tanto clínico como quirúrgico. Por lo tanto, se presenta un caso de un paciente con ICE que pasó por consultas previas en otros servicios sin haber tenido un correcto diagnóstico temprano.
Caso clínico
Paciente de sexo femenino de 42 años con antecedente de disminución de la agudeza visual (AV) en OI, siendo su AV mejor corregida en ojo derecho (OD) de 10/10 y en ojo izquierdo (OI) de 8/10, con una presión intraocular (PIO) en OD: 17 mmHg y en OI: 35 mmHg, con tratamiento hipotensor ocular máximo. Presentaba al examen del segmento anterior córnea transparente con imagen de cobre martillado en el endotelio corneal, con pupila corectópica, atrofia del iris difusa y ectropión uveal (fig. 1); en la gonioscopía mostró sinequias anteriores periféricas altas y de base ancha que se insertaban a nivel de malla trabecular y línea de Schwalbe con cierre angular secundario (fig. 2).
Al examen del nervio óptico del OD se observó relación copa/disco fisiológica y en OI, importante atrofia del anillo neurorretinal con aumento de la excavación papilar. Se le realizó tomografía de coherencia óptica y campimetría computarizada donde se constató respectivamente pérdida de la capa de fibras nerviosas peripapilares en OI, sin alteraciones en OD y defectos campimétricos arqueados en OI. En la microscopía especular se observó disminución en el recuento de células endoteliales con polimegatismo y pleomorfismo asociado, así como aumento del espesor corneal (fig. 3). Por los datos anteriores se arribó al diagnóstico de ICE. Y, ante los elevados valores de PIO, se propuso y se sometió a la paciente a una cirugía de implante valvular con dispositivo de drenaje de Ahmed (fig. 4). A los seis meses del procedimiento quirúrgico mantuvo la PIO en 12 mmHg sin medicación tópica hipotensora asociada, con córnea transparente sin progresión estructural ni funcional.
DISCUSIÓN
El síndrome de ICE es una patología poco frecuente en la cual existe una proliferación endotelial corneal que migra hacia el ángulo iridocorneal y sobre el iris con diferentes grados de edema corneal, alteraciones iridianas y cierre angular progresivo con el consiguiente desarrollo de glaucoma1-4.
Aunque aún no está bien fundamentada la patogenia, la más aceptada es la relacionada a una infección viral5. El hecho de que suele ser unilateral puede explicarse por el desarrollo de anticuerpos, los cuales evitarían el daño al ojo contralateral en la gran mayoría de los casos. Se ha informado que el ADN viral del herpes simple se ha recuperado de la córnea de pacientes ICE, pero su papel patológico es aún especulativo5. En este caso, la paciente negaba todo tipo de antecedentes de infecciones virales a nivel ocular y no se realizaron pruebas para evaluar esta etiología como podría ser la PCR de humor acuoso.
Figura 1. Segmento anterior del OI con córnea transparente, imagen de cobre martillado en el endotelio, pupila corectópica, atrofia del iris difusa y ectropión uveal.

Figura 2. Gonisocopía en OI con presencia de sinequias anteriores periféricas altas y de base ancha que se insertan a nivel de malla trabecular y línea de Schwalbe con cierre angular secundario.
En su expresión clínica presenta tres variantes: el síndrome de Chandler (CS, por sus siglas en inglés), atrofia iridiana progresiva (PIA, por sus siglas en inglés) y el síndrome de Cogan-Reese (CRS, por sus siglas en inglés)2-3. La variante CS ha sido descrita como la más frecuente en blancos y en asiáticos; CRS se informó como la forma más común y es fuertemente asociado con el glaucoma6.
La prevalencia de glaucoma en estos pacientes ronda entre el 46% y el 82%6. La gravedad del glaucoma se constata más avanzada en CRS y PIA en comparación con CS, el cual se atribuye a la mayor prevalencia de proliferación de células ICE sobre el ángulo iridocorneal y la presencia de PAS que se presentan en estas variantes7.
El control de la PIO se considera un desafío para pacientes con SRC o PIA que en aquellos con SC, independientemente de la extensión de PAS. Una serie publicada por Wilson y Shields describió mayor proporción de la necesidad de cirugía con dispositivos filtrantes en pacientes con SRC (50%) o PIA (75%) en comparación con CS (40%)8.

Figura 3. Microscopía especular de OI. Se observa disminución en el recuento de células endoteliales con polimegatismo y pleomorfismo asociado y aumento del espesor corneal.
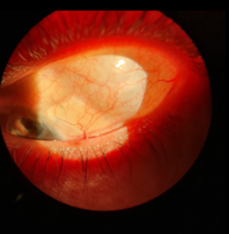
Figura 4. Implante de válvula de Ahmed en OI. Se observa tubo en cámara anterior, ampolla formada, elevada y difusa.
CONCLUSIÓN
Finalmente y a modo de conclusión, ante la sospecha de un ICE es importante realizar un examen completo del segmento anterior, buscando signos en el iris como en la córnea para realizar un correcto diagnóstico. En todos los pacientes resulta elemental el seguimiento mediante exploraciones periódicas para poder actuar de forma precoz ante el inicio de complicaciones.
Referencias
1. Eagle RC Jr., Font RL, Yanoff M, Fine BS. Proliferative endotheliopathy with iris abnormalities: the iridocorneal endothelial syndrome. Arch Ophthalmol 1979; 97: 2104-2111.
2. Walkden A, Au L. Iridocorneal endothelial syndrome: clinical perspectives. Clin Ophthalmol 2018; 12: 657-664.
3. Alvarado JA, Murphy CG, Maglio M, Hetherington J. Pathogenesis of Chandler’s syndrome, essential iris atrophy and the Cogan-Reese syndrome. I. Alterations of the corneal endothelium. Invest Ophthalmol Vis Sci 1986; 27: 853-872.
4. Sacchetti M, Mantelli F, Marenco M et al. Diagnosis and Management of iridocorneal endothelial syndrome. Biomed Res Int 2015; 2015: 763093.
5. Alvarado JA, Underwood JL, Green WR et al. Detection of herpes simplex viral DNA in the iridocorneal endothelial syndrome. Arch Ophthalmol 1994; 112: 1601-1609.
6. Teekhasaenee C, Ritch R. Iridocorneal endothelial syndrome in Thai patients: clinical variations. Arch Ophthalmol 2000; 118: 187-192.
7. Laganowski HC, Kerr Muir MG, Hitchings RA. Glaucoma and the iridocorneal endothelial syndrome. Arch Ophthalmol 1992; 110: 346-350.
8. Wilson MC, Shields MB. A comparison of the clinical variations of the iridocorneal endothelial syndrome. Arch Ophthalmol 1989; 107: 1465-1468.